|
|
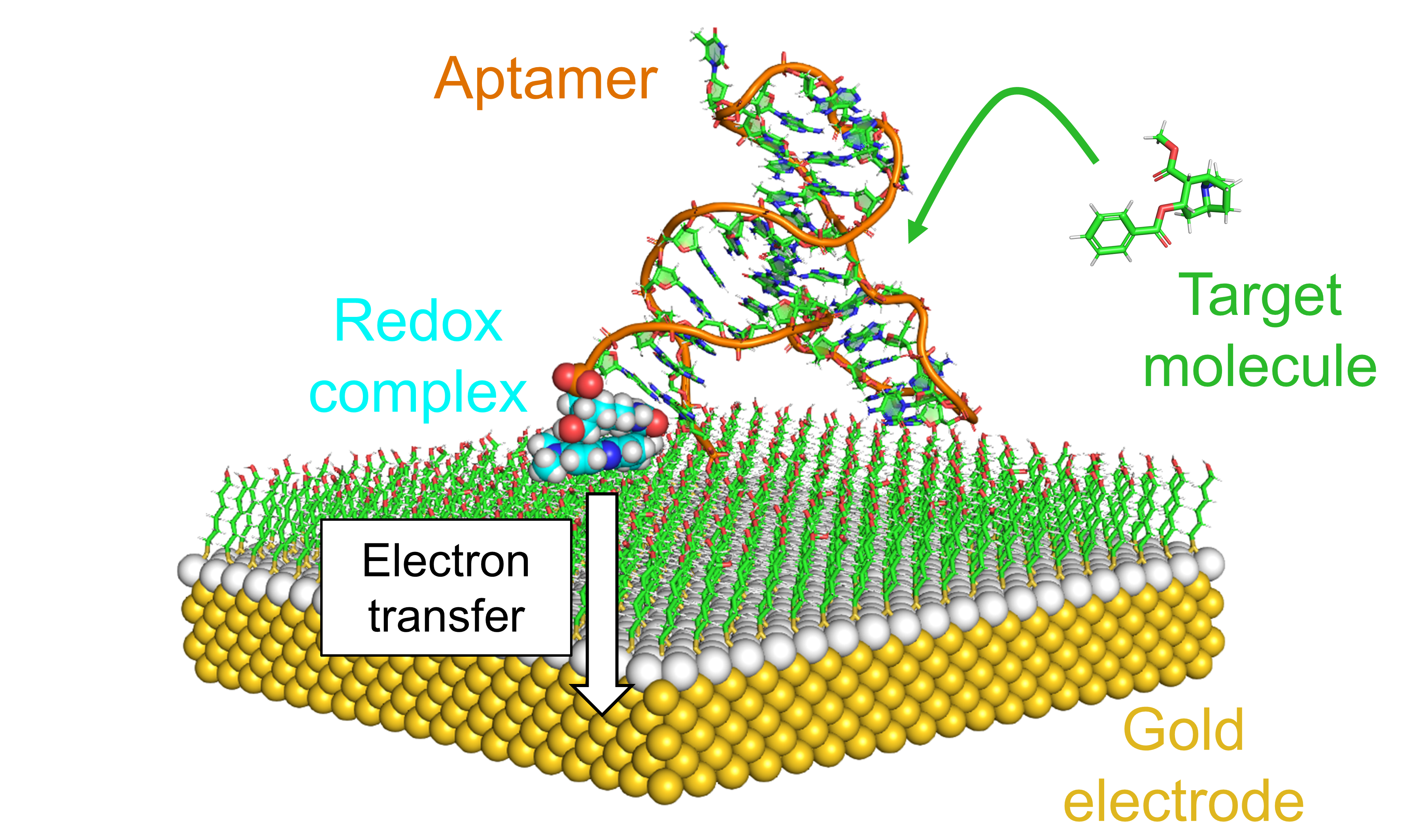
Bioelectrochemical devices rely on a transducer surface bridging their electrical circuit and the environment in which they operate. Electrochemical biosensors, for example, can use a gold electrode functionalized with aptamers to detect small molecules in a biological sample (see Figure) [1]. Specifically, binding of the target molecule induces conformational changes in the aptamer that, in turns, affect the electron transfer between its redox complex and the electrode. As seen in this example, the biomolecule-transducer interface needs to be designed precisely to achieve optimal operation because the electronic signal of the device is driven by the nanoscale interactions at that interface. This is where simulations come into play as they unveil with a nanoscale resolution the interactions between the biomolecules and the transducer surface [2,3]. These atomistic insights guide the design of optimal bioelectrochemical devices.
Simulations consist of two parts: modeling and sampling. In molecular mechanics models, the chemical structure is fixed, and each atom is treated as single particle interacting through bonded (covalent bonds, bond angles and torsion angles) and non-bonded (van der Waals and electrostatic) terms with the other atoms. When modeling the biomolecule-transducer interface, models are chosen for the biomolecules and for the transducer surface. While all-atom models for biomolecules (proteins, nucleic acids, etc.) in water such as AMBER and CHARMM are well established, models for transducer surfaces (metal, semiconductor, polymers, etc.) are continuously developed as new types of transducer surfaces are envisioned, and their compatibility with biomolecules needs to be assessed [4,5]. Once the models are chosen, a sampling algorithm is used to unveil the quantities of interest such as the preferred biomolecule-transducer interactions, the conformational changes in the biomolecule and the energy profile of the biomolecule-transducer association. Molecular dynamics is the most used technique whereby Newton’s equation of motion is iteratively solved to get the dynamics of the system at constant temperature and pressure. When sampling the biomolecule-transducer interface, the dynamics should span long enough timescales to reach ergodicity, depending on the questions addressed. Often, however, microsecond-long simulations are not sufficient and techniques that instead sample the thermodynamics of the system should be used, such as replica-exchange, metadynamics or umbrella sampling; or it is also possible to use implicit solvent and coarse-grained models [6,7].
As seen in the cited reviews, simulating biomolecules at transducer surfaces is an exciting research field where meet the development of force field models and sampling techniques to characterize a wealth of biomolecule-transducer interfaces.
|
|
|
References:
[1] Downs, A. M.; Plaxco, K. W. Real-time, in vivo molecular monitoring using electrochemical aptamer-based sensors: opportunities and challenges. ACS Sens. 2022, 7, 2823-2832.
[2] Ozboyaci, M.; Kokh, D. B.; Corni, S.; Wade, R. C. Modeling and simulation of protein-surface interactions: achievements and challenges. Q. Rev. Biophys. 2016, 49, e4, 1-45.
[3] Dutta, S.; Corni, S.; Brancolini, G. Atomistic simulations of functionalized nano-materials for biosensors applications. Int. J. Mol. Sci. 2022, 23, 1484.
[4] Dasetty, S.; Meza-Morales, P. J.; Getman, R. B.; Sarupria, S. Simulations of interfacial processes: recent advances in force field development. Curr. Opin. Chem. Eng. 2019, 23, 138-145.
[5] Choi, Y. K.; Kern, N. R.; Kim, S.; Kanhaiya, K.; Afshar, Y.; Jeon, S. H.; Jo, S.; Brooks, B. R.; Lee, J.; Tadmor, E. B.; Heinz, H.; Im, W. CHARMM-GUI nanomaterial modeler for modeling and simulation of nanomaterial systems. J. Chem. Theo. Comput. 2022, 18, 479-493.
[6] Quan, X.; Liu, J.; Zhou, J. Multiscale modeling and simulations of protein adsorption: progresses and perspectives. Cur. Opin. Colloid Interf. Sci. 2019, 41, 74-85.
[7] Lbadaoui-Darvas, M.; Garberoglio, G.; Karadima, K. S.; Cordeiro, M. N. D. S.; Nenes, A.; Takahama, S. Molecular simulations of interfacial systems: challenges, applications and future perspectives. Mol. Simulat. 2023, 49, 1229-1266.
|
|